
绘制热图除了使用ggplot2,还可以有其它的包或函数,比如pheatmap::pheatmap (pheatmap包中的pheatmap函数)、gplots::heatmap.2等。
相比于ggplot2作heatmap, pheatmap会更为简单一些,一个函数设置不同的参数,可以完成行列聚类、行列注释、Z-score计算、颜色自定义等。
data_ori <- "Grp_1;Grp_2;Grp_3;Grp_4;Grp_5 a;6.6;20.9;100.1;600.0;5.2 b;20.8;99.8;700.0;3.7;19.2 c;100.0;800.0;6.2;21.4;98.6 d;900;3.3;20.3;101.1;10000" data <- read.table(text=data_ori, header=T, row.names=1, sep=";", quote="") Grp_1 Grp_2 Grp_3 Grp_4 Grp_5 a 6.6 20.9 100.1 600.0 5.2 b 20.8 99.8 700.0 3.7 19.2 c 100.0 800.0 6.2 21.4 98.6 d 900.0 3.3 20.3 101.1 10000.0 pheatmap::pheatmap(data, filename="pheatmap_1.pdf")
虽然有点丑,但一步就出来了。
此外Z-score计算在pheatmap中只要一个参数就可以实现。
pheatmap::pheatmap(data, scale="row", filename="pheatmap_1.pdf")
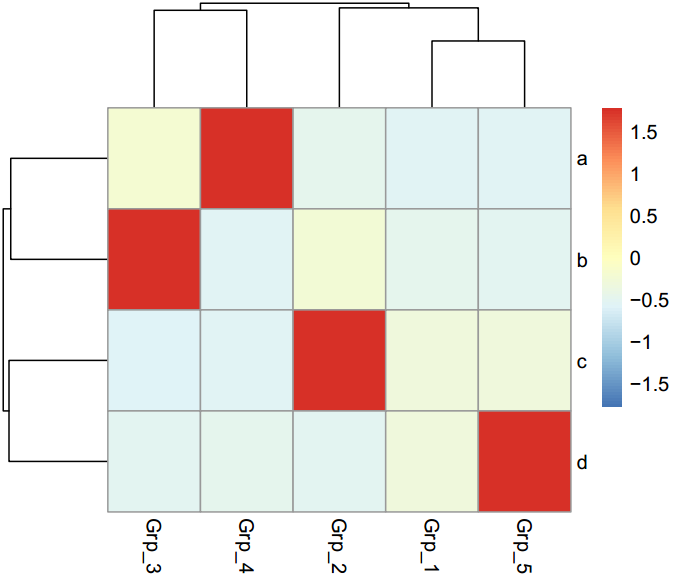
有时可能不需要行或列的聚类,原始展示就可以了。
pheatmap::pheatmap(data, scale="row", cluster_rows=FALSE, cluster_cols=FALSE, filename="pheatmap_1.pdf")
给矩阵 (data)中行和列不同的分组注释。假如有两个文件,第一个文件为行注释,其第一列与矩阵中的第一列内容相同 (顺序没有关系),其它列为第一列的不同的标记,如下面示例中(假设行为基因,列为样品)的2,3列对应基因的不同类型 (TF or enzyme)和不同分组。第二个文件为列注释,其第一列与矩阵中第一行内容相同,其它列则为样品的注释。
row_anno = data.frame(type=c("TF","Enzyme","Enzyme","TF"), class="c"("clu1","clu1","clu2","clu2"), row.names=rownames(data))
row_anno
type class
a TF clu1
b Enzyme clu1
c Enzyme clu2
d TF clu2
col_anno = data.frame(grp=c("A","A","A","B","B"), size=1:5, row.names=colnames(data))
col_anno
grp size
Grp_1 A 1
Grp_2 A 2
Grp_3 A 3
Grp_4 B 4
Grp_5 B 5
pheatmap::pheatmap(data, scale="row",
cluster_rows=FALSE,
annotation_col=col_anno,
annotation_row=row_anno,
filename="pheatmap_1.pdf")
自定义下颜色吧。
# <bias> values larger than 1 will give more color for high end.
# Values between 0-1 will give more color for low end.
pheatmap::pheatmap(data, scale="row",
cluster_rows=FALSE,
annotation_col=col_anno,
annotation_row=row_anno,
color=colorRampPalette(c('green','yellow','red'), bias=1)(50),
filename="pheatmap_1.pdf")
不改脚本的热图绘制
绘图时通常会碰到两个头疼的问题:
- 需要画很多的图,唯一的不同就是输出文件,其它都不需要修改。如果用R脚本,需要反复替换文件名,繁琐又容易出错。
- 每次绘图都需要不断的调整参数,时间久了不用,就忘记参数放哪了;或者调整次数过多,有了很多版本,最后不知道用哪个了。
为了简化绘图、维持脚本的一致,我用bash对R做了一个封装,然后就可以通过修改命令参数绘制不同的图了。
先看一看怎么使用
首先把测试数据存储到文件中方便调用。数据矩阵存储在heatmap_data.xls文件中;行注释存储在heatmap_row_anno.xls文件中;列注释存储在heatmap_col_anno.xls文件中。
# tab键分割,每列不加引号
write.table(data, file="heatmap_data.xls", sep="t", row.names=T, col.names=T, quote=F)
# 如果看着第一行少了ID列不爽,可以填补下。-i参数直接对文件进行操作,1 指定第一行。在行首添加制表符
system("sed -i '1 s/^/IDt/' heatmap_data.xls")
write.table(row_anno, file="heatmap_row_anno.xls", sep="t", row.names=T, col.names=T, quote=F)
write.table(col_anno, file="heatmap_col_anno.xls", sep="t", row.names=T, col.names=T, quote=F)
然后用程序sp_pheatmap.sh绘图。
# -f: 指定输入的矩阵文件 # -d:指定是否计算Z-score,<none> (否), <row> (按行算), <col> (按列算) # -P: 行注释文件 # -Q: 列注释文件 $ sp_pheatmap.sh -f heatmap_data.xls -d row -P heatmap_row_anno.xls -Q heatmap_col_anno.xls
一个回车就得到了图,字有点小,是因为图太大了,把图的宽和高缩小下试试。
# -u: 设置宽度,单位是inch
# -v: 设置高度,单位是inch
$ sp_pheatmap.sh -f heatmap_data.xls -d row -P heatmap_row_anno.xls -Q heatmap_col_anno.xls -u 8 -v 12
横轴的标记水平放置
# -A: 0, X轴标签选择0度
# -C: 自定义颜色,注意引号的使用,最外层引号与内层引号不同,引号之间无交叉
# -T: 指定给定的颜色的类型;如果给的是vector (如下面的例子), 则-T需要指定为vector; 否则结果会很怪异,只有俩颜色。
# -t: 指定图形的题目,注意引号的使用;参数中包含空格或特殊字符等都要用引号引起来作为一个整体。
$ sp_pheatmap.sh -f heatmap_data.xls -d row -P heatmap_row_anno.xls -Q heatmap_col_anno.xls -u 8 -v 12 -A 0 -C 'c("white", "blue")' -T vector -t "Heatmap of gene expression profile"
内容来源于网络如有侵权请私信删除
- 还没有人评论,欢迎说说您的想法!


 客服
客服


